[Frontier Information] CRISPRi Screening Boosts the Discovery of Key Targets in Tumor Immunity

Tumor immunotherapy is a therapeutic approach that harnesses immunological principles and techniques to activate endogenous immune cells and amplify the body's antitumor immune response, specifically targeting and eliminating residual tumor cells while suppressing tumor growth. It aims to counteract the mechanisms of immune evasion employed by tumors and reinvigorate immune cells for cancer cell clearance. With its reduced side effects and promising efficacy, immunotherapy is emerging as a key frontier in cancer treatment, complementing surgery, radiation, and chemotherapy as the fourth major modality.
In May, researchers from Harvard Medical School published a study titled "Targetable leukemia dependency on noncanonical PI3Kγ signaling" in Nature, with an impact factor of 64.8. The study highlights that PI3Kγ suppresses tumor-associated macrophages' stimulation of antitumor immunity, making it a crucial target for immunotherapy. However, PI3Kγ inhibitors have shown limited antitumor efficacy. The researchers employed CRISPR interference (CRISPRi) screening, combined with unbiased genome-wide screening and functional analysis, to identify gene dependencies in the BPDCN cell line. They discovered that p21-activated kinase 1 (PAK1) acts as an atypical substrate in the non-canonical PI3Kγ signaling pathway, mediating intrinsic cellular dependence. This finding suggests that inhibiting PAK1 could suppress its phosphorylation, thereby overcoming resistance to PI3Kγ inhibitors in lymphoma, ultimately enhancing the effectiveness of tumor immunotherapy.

Acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) are the most common acute leukemias in adults and children, respectively, while blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare and aggressive hematological malignancy originating from plasmacytoid dendritic cell (pDC) lineage, sharing clinical and pathological features with AML and ALL. The mammalian phosphoinositide 3-kinase (PI3K) family encompasses eight isoforms, with Class IA and IB PI3Ks generating three phosphoinositide lipids to activate signaling pathways, while Class II and III PI3Ks regulate membrane trafficking along endocytic pathways. Class IA PI3K inhibitors have been approved or are under development, whereas Class IB PI3Ks, particularly PI3Kγ inhibitors, have garnered less attention, primarily for reprogramming macrophages in solid tumor immunotherapy. Researchers conducted whole-genome CRISPRi screening and integrated analysis to define a non-canonical PI3Kγ signaling dependency shared by most BPDCNs and other acute leukemias (AMLs and ALLs). PI3Kγ signaling, through PIK3R5 activation and phosphorylation of p21-activated kinase 1 (PAK1), was found to drive the malignant phenotype and showed sensitivity to PI3Kγ inhibition, offering a novel therapeutic strategy for aggressive leukemias.
I. Integrating CRISPRi and bioinformatics screens reveals intrinsic dependence of leukemia on the PI3Kγ complex
Utilizing the BPDCN cell line CAL-1, researchers performed whole-genome CRISPRi dependency screens with Dolcetto guide RNAs from groups A and B. Samples were collected at days 0, 14, and 21 following puromycin selection, and as expected, the highest dependencies at days 14 and 21 corresponded to ribosomal proteins and cancer dependencies from the DepMap. Among 1335 significant CAL-1 dependencies, 327 genes were defined as core dependencies with a stringent threshold of P < 0.001 and fold change < 0.5 at day 21. No genes overlapped with DepMap's non-essential negative control genes at day 21, while 143 of the 327 core dependencies were also common factors in DepMap, indicating 184 genes as potential specific CAL-1/BPDCN dependencies.
To address the scarcity of BPDCN cell lines, researchers modified an established organoid culture protocol, enabling primary patient samples to survive in vitro for at least two weeks, developing a leukemia-adapted medium (LAM) that successfully generated new BPDCN cell lines from patient samples. Additionally, various PI3K inhibitors were tested, and results consistently showed that the three leukemia cell lines with the highest PIK3R5/PIK3CG expression were highly sensitive to the selective PI3Kγ inhibitor eganelisib, suggesting that PIK3R5/PIK3CG expression serves as a specific marker of PI3Kγ dependence.
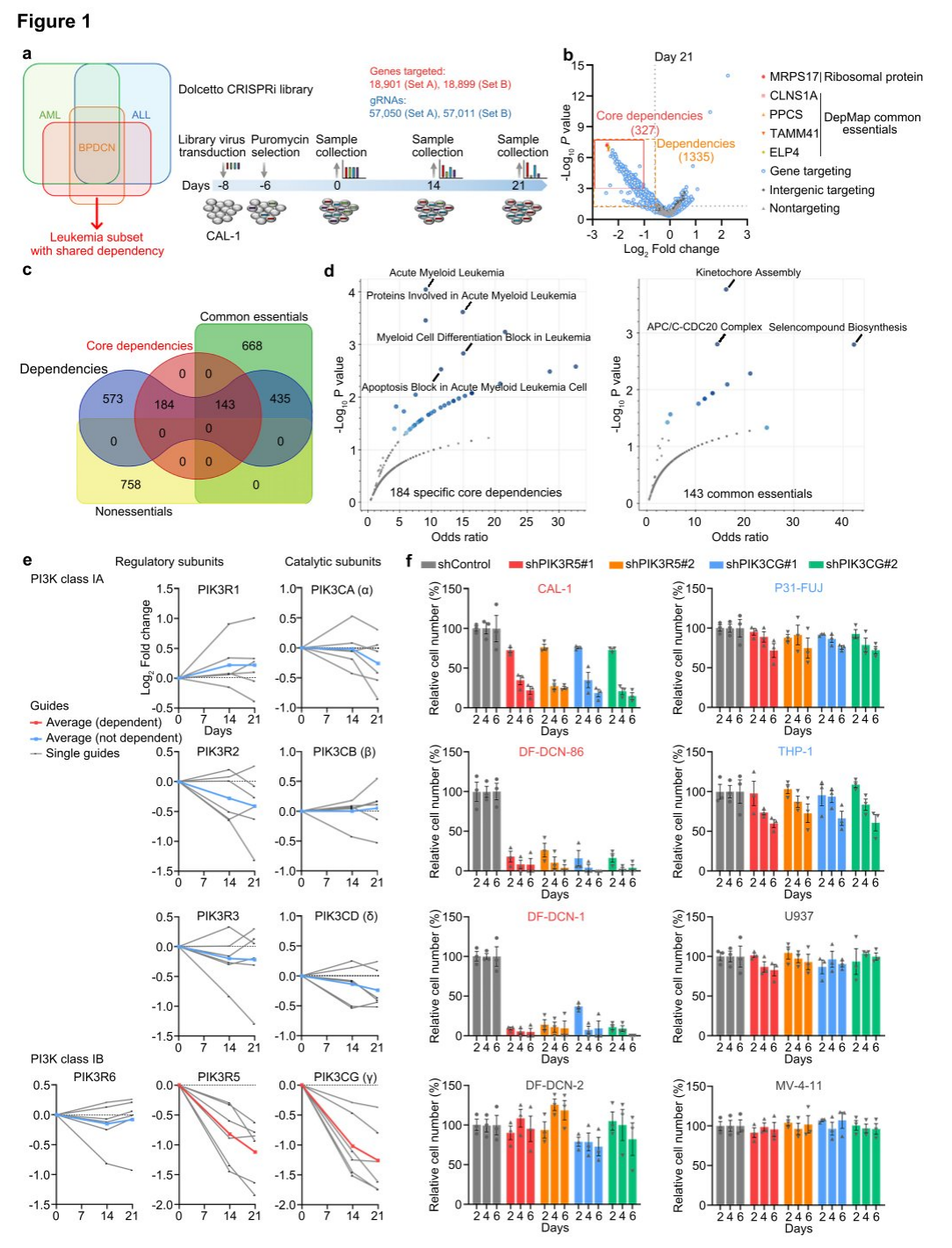
Figure 1: Genome-wide CRISPRi screening identifies selective dependence of BPDCN on PI3Kγ.
II. PIK3R5 expression is associated with the Innate Immune Response Signature (IIRS) and can be activated by innate inflammatory signals.
To further establish the dependency on the PI3Kγ complex, researchers assessed the vulnerability of AML, ALL, and BPDCN patient-derived xenograft (PDX) models to PI3Kγ inhibition. Leukemias with elevated PIK3R5 expression were sensitive to eganelisib, while those with low PIK3R5 expression were not, indicating a shared intrinsic dependency on the PI3Kγ complex across various leukemia subtypes, which can be predicted by PIK3R5 mRNA expression levels.
To further characterize leukemias with increased PIK3R5 expression, a gene set enrichment analysis (GSEA) was conducted, comparing cases with high and low PIK3R5 expression. All top-enriched gene sets were related to innate immune response pathways. After excluding BPDCN cases from the analysis to control for potential biases due to the dendritic cell origin of BPDCN, the same conclusion was reached. A multivariate analysis of clinical and pathological factors in the TCGA cohort revealed a significant association between IIRS scores and poor prognosis, with no specific enrichment of recurrent mutations in the positive group, suggesting that PIK3R5 expression and IIRS scores predict PI3Kγ dependency and may define leukemia subgroups related to inflammation.
To further investigate the relationship between innate inflammatory signals and PIK3R5 expression, researchers treated leukemia cell lines with Resiquimod. The treatment significantly increased PIK3R5 expression in leukemia cells with low baseline levels, without altering PIK3CG mRNA levels across all cell lines. However, PIK3CG protein levels rose in parallel with PIK3R5, indicating that activation of innate inflammatory signals contributes to the upregulation of PIK3R5 expression and enhances sensitivity to eganelisib.
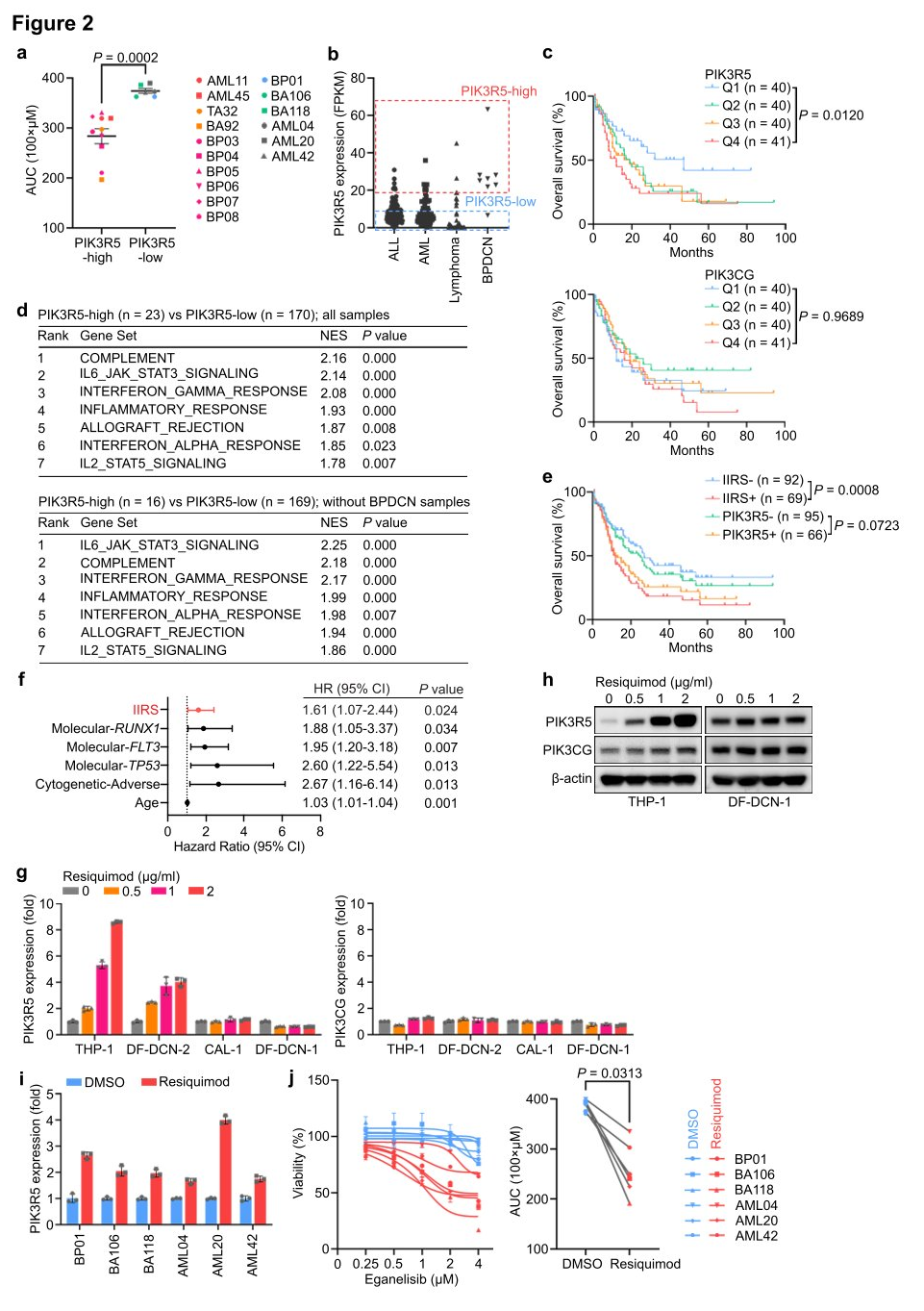
Figure 2: A subset of leukemia defined by activation of PIK3R5 by innate inflammatory signals exhibits selective dependence on the PI3Kγ subunit.
III. PU.1 activates PIK3R5, promoting self-stabilization of the PI3Kγ complex.
Researchers utilized high-throughput sequencing (ATAC-seq) to examine the transposase and chromatin of eight AML, ALL, and BPDCN PDX models to identify upstream transcriptional activators that enhance PIK3R5 expression and dependency on the PI3Kγ complex. The measurements revealed that cells with high levels of PIK3R5 had more accessible chromatin regions around the PIK3R5 transcription start site (TSS) compared to cells with low PIK3R5 levels, regardless of leukemia histology. Analysis of Chromatin Immunoprecipitation and sequencing (ChIP-seq) data from the ENCODE database identified 34 transcriptional regulatory factors interacting near the PIK3R5 promoter TSS. Among the genes encoding these 34 transcriptional regulatory factors, SPI1 and ASH2L were core dependency genes in the genome-wide CRISPRi screen. Therefore, researchers hypothesized that one of these transcriptional regulators might control the activation of PIK3R5.
Researchers knocked out SPI1 and ASH2 genes separately and measured the effect on PIK3R5 expression. The results showed that knocking out the SPI1 gene significantly reduced the levels of PIK3R5 mRNA and protein. SPI1 encodes the transcription factor PU.1, which plays a crucial role in myeloid and lymphoid cell differentiation and maintenance of hematopoietic stem cells. ChIP analysis observed an increased association between PU.1 and the PIK3R5 promoter after Resiquimod treatment, indicating that PU.1 is a key transcription factor mediating PIK3R5 activation in PI3Kγ-dependent leukemia subgroups. Knocking out the ASH2 gene decreased the protein level of PIK3CG, but the absence of PIK3R5 or PIK3CG did not affect the mRNA expression of each other. Researchers suggested that PIK3R5 and PIK3CG proteins might mutually protect each other, thereby increasing their half-life in cells. To verify this possibility, researchers treated CAL-1 cells with the protein synthesis inhibitor emetine and monitored the stability of PIK3R5 and PIK3CG proteins. The results showed that the absence of PIK3CG significantly reduced the stability of the PIK3R5 protein, and vice versa. Ubiquitination experiments found that the absence of PIK3R5 or PIK3CG enhanced the ubiquitination of the other, further supporting the hypothesis of this shared protein protection mechanism.
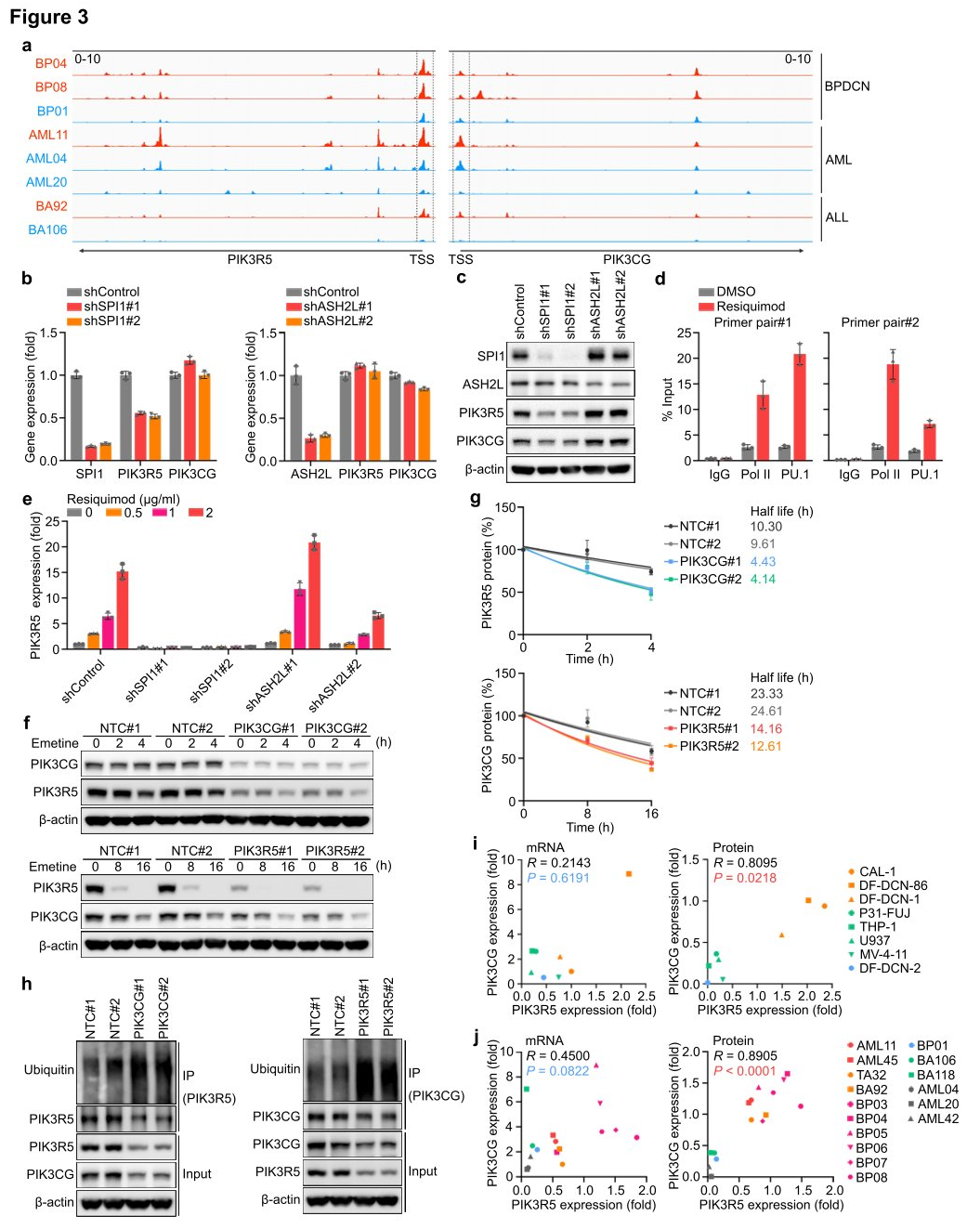
Figure 3: PIK3R5 is transcriptionally activated by PU.1 and protects the PI3Kγ complex from degradation mediated by the UPS (Ubiquitin-Proteasome System).
IV. PI3Kγ inhibits OXPHOS and activates an NFkB-related transcriptional network.
To investigate the downstream events dependent on intrinsic PI3Kγ in leukemia cells, researchers conducted integrated RNA sequencing, proteomics, and phosphoproteomics analyses using CAL-1 cells treated with eganelisib or CAL-1 cells where PIK3R5 was removed using CRISPRi (PIK3R5i). In both inhibition and depletion scenarios, GSEA of the cell transcriptome revealed that the most significantly downregulated molecular event following targeting of PI3Kγ was associated with OXPHOS, providing additional rationale for selectively targeting mitochondria in leukemia cells through PI3Kγ inhibition. After treatment with eganelisib, 11 genes (RHOB, EGR1, CD69, SGK1, and EGR3) were consistently upregulated, with five located within the core enrichment group of this gene set, several of which have tumor suppressive roles in various cancers. To verify whether one or more of these core enriched genes could lead to sensitivity to PI3Kγ inhibition in PIK3R5-related leukemia subtypes, validation was performed in CAL-1 and DF-DCN-1 cells. Knockout of four out of the five genes reduced sensitivity to PI3Kγ inhibition. Treatment of CAL-1 cells with selective NFkB inhibitors IKK-16 and SC-514 found that eganelisib no longer increased the expression of these five genes. Metabolic analysis showed that NFkB inhibition blocked the suppression of OXPHOS by PI3Kγ inhibitors, and CAL-1 cells treated with NFkB inhibitors became resistant to eganelisib. The study data indicate that PI3Kγ dependency is partially mediated by an NFkB-related transcriptional network.
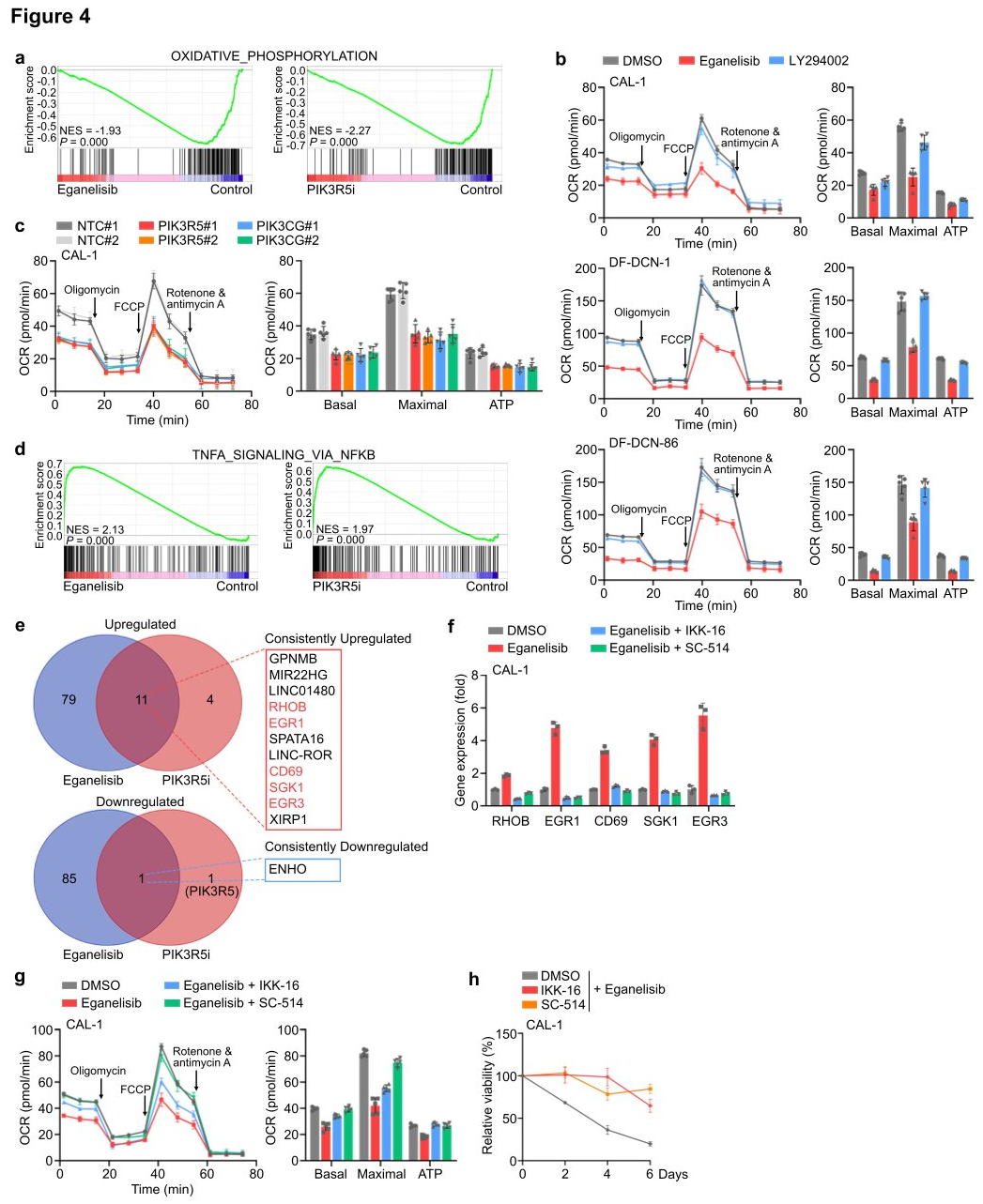
Figure 4: PI3Kγ inhibition activates an NFkB-related transcriptional network and suppresses OXPHOS in leukemia cells.
V. PI3Kγ inhibition does not rely on the atypical PI3K pathway independent of Akt kinase (Akt)
Researchers found through phosphoproteomic measurements that after PI3Kγ inhibition, there was no significant change in AKT phosphorylation in CAL-1 cells. GSEA also revealed that the classical PI3K-AKT-mTOR signaling target genes remained unchanged following treatment with eganelisib or upon PIK3R5 depletion, suggesting that atypical PI3K mediators might contribute to the dependency on PI3Kγ in PIK3R5-related leukemia subtypes. Further analysis of proteomic and phosphoproteomic data identified several proteins whose phosphorylation was affected by PI3Kγ inhibition and PIK3R5 deletion. Western blotting confirmed that phosphorylation at serine 144 (S144) of PAK1 was significantly reduced following PIK3R5/PIK3CG depletion, while phosphorylation of other known proteins in the typical PI3K signaling pathway was unaffected. Notably, the absence of PIK3R5 or PIK3CG led to a complementary increase in AKT phosphorylation, whereas eganelisib induced suppression of AKT phosphorylation.
To further verify the functional involvement of PAK1 S144 phosphorylation in PI3Kγ dependency, researchers treated CAL-1 cells with selective inhibitors of PAK1 or AKT. Treatment with a PAK1 inhibitor mimicked the strong inhibition of CAL-1 growth observed with PI3Kγ inhibition, while the AKT inhibitor MK-2206 completely abolished AKT phosphorylation but only slightly impaired cell viability. The introduction of exogenous constitutively active (S144D) or inactive (S144A) PAK1 phosphorylation site mutants into CAL-1 cells, followed by treatment with eganelisib, conferred resistance to PI3Kγ inhibition. Finally, researchers tested the impact of PAK1 S144 phosphorylation on downstream events of PI3Kγ. Hippocampal experiments showed that the introduction of PAK1 S144D eliminated the suppression of OXPHOS by PI3Kγ inhibition, and the elevation of NFkB-related tumor suppressor genes induced by eganelisib was also reduced in CAL-1 cells expressing PAK1 S144D, indicating that PAK1 is a key substrate of PI3Kγ in the leukemia-intrinsic dependency pathway, with PAK1 S144 phosphorylation playing a crucial role in this signaling pathway.
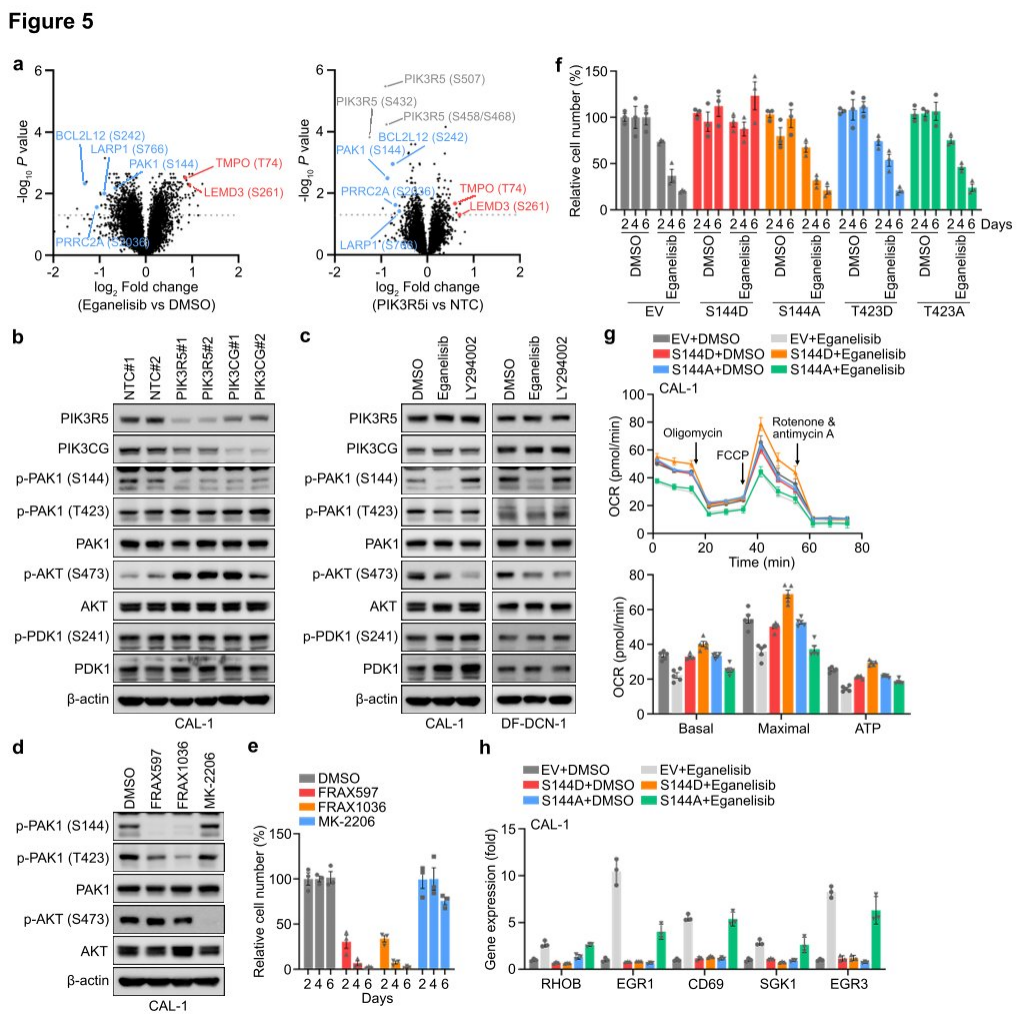
Figure 5: Leukemia cells rely on PAK1 for their intrinsic dependence on the PI3Kγ complex.
VI. Synergistic Treatment of Leukemia with PI3Kγ Inhibition and Cytarabine
To explore the translational potential of targeting PI3Kγ in leukemia, researchers tested the combination of eganelisib with cytarabine in two BPDCN cell lines with elevated PIK3R5, observing a significant synergistic effect in both cell lines. Next, they evaluated the dynamic response of leukemia cells in vivo using the DF-DCN-86 subcutaneous xenograft model. The results showed that eganelisib was more effective as a single agent compared to cytarabine, and the combination of both drugs synergistically inhibited tumor growth. Western blot analysis of tumor lysates harvested immediately after treatment in animals treated with eganelisib or eganelisib plus cytarabine revealed reduced PAK1S144 phosphorylation, but no decrease in T423 phosphorylation. Lastly, the researchers measured the impact of PI3Kγ inhibition on survival in six disseminated PDX models. The results demonstrated that single-agent eganelisib significantly prolonged survival in AML, ALL, and BPDCN patients with high PIK3R5 levels, with no benefit observed in patients with low PIK3R5 expression.
Based on the in vivo efficacy, researchers sought to understand why the combination of eganelisib and cytarabine was synergistic even in leukemias without elevated PIK3R5. They treated three PDX models with low PIK3R5 expression in vitro with cytarabine concentrations that resulted in approximately 20% persistent leukemia cells for analysis of residual disease. The results showed that the cytarabine-resistant cells exhibited significantly increased PAK1S144 phosphorylation without changes in AKT S473, which might explain their induced sensitivity to eganelisib. The data suggest that the G protein-coupled purinergic receptor-PI3Kγ-PAK1 signaling pathway activated in cytarabine-resistant leukemia may also be targeted by PI3Kγ inhibition, expanding the potential clinical application of PI3Kγ inhibitors in leukemias without baseline PIK3R5 activation.
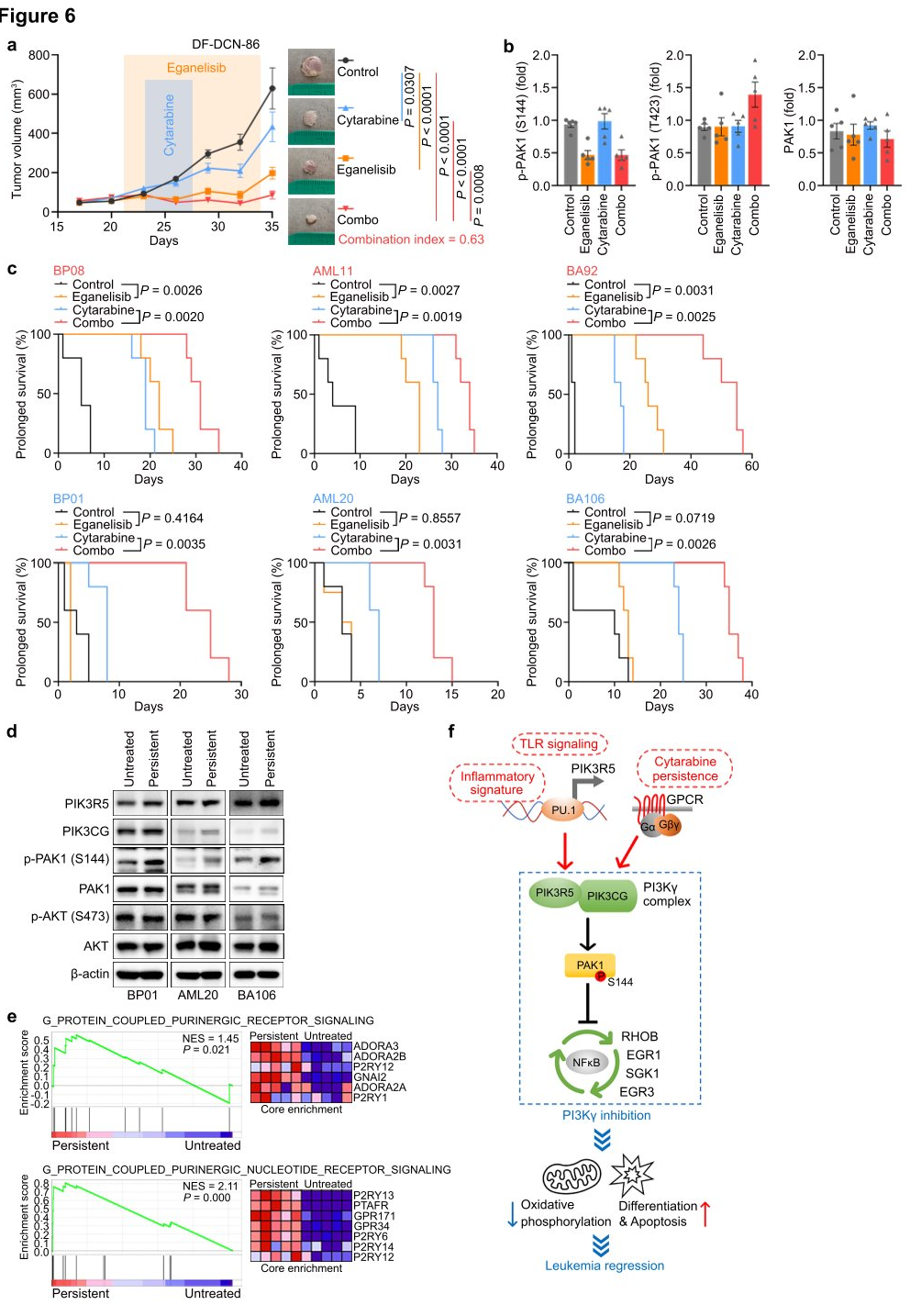
Figure 6: Synergy between Eganelisib and Cytarabine in Treating Leukemia
In summary, researchers utilized the CRISPRi screening technology combined with unbiased genome-wide screening and functional analysis to identify selective dependencies on the PI3Kγ complex within high-risk subgroups of BPDCN (plasmacytoid dendritic cell neoplasm) and other acute leukemias, including AML and ALL. The study revealed p21 (RAC1) activated kinase 1 (PAK1) as an atypical substrate for PI3Kγ, providing a new therapeutic target for acute leukemia.
Original Article Link: https://doi.org/10.1038/s41586-024-07410-3
EDITGENE offers a one-stop complete solution for CRISPR library screening, providing CRISPR KO/CRISPRa/CRISPRi services, along with the most comprehensive domestic collection of plasmid/virus libraries available for immediate delivery within a week upon order placement.
Recent Blogs
1.【Cutting-Edge News】New Trends in Gene Editing - FGFR1, BRD4, and BACE1 Knockout Cells
2.A talk about the Past and Present of Gene Editing Technology: ZFN, TALEN and CRISPR
Follow us on social media





Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com